Graph-based gene prediction with ggCaller
By Sam Horsfield
In this blog, I give a brief overview of bacterial pangenome analysis, and what problems our tool, ggCaller, solves.
The bacterial pangenome – quantifying within-species diversity
A genome is a set of biological instructions, known as ‘genes’, which describe how to make and maintain a living organism. The biological similarities and differences we observe between organisms, either between those that are closely or distantly related, are in no small part due to the genes they possess. By identifying the genes within an organism’s genome, we can make predictions about how it behaves, such as how it interacts with other organisms, and in which environments it can survive.
In bacteria, we observe a great diversity of biological characteristics even between closely related individuals, i.e. those belonging to the same species. By identifying and comparing the genes that are shared and different between individuals, we can characterise which genes define certain characteristics. We might want to do this to understand how a strain is resistant to antibiotics, why it is more pathogenic, or why it is able to evade vaccines.
Comparing the genes within a population represents a ‘pangenome’ – the collection of all the unique genes. Almost universally across species, we observe a large number of common or ‘core’ genes which are found in many individuals, as well as large number of rare or ‘accessory’ genes, which are only found in certain individuals. Core genes define the fundamental characteristics of a species, whilst accessory genes define the diversity we see between individuals.
How do you represent a pangenome?
To generate a representation of a pangenome, we first need to identify all of the genes in all of the genomes we have for a given population. After that, we need to compare each gene against all others to identify which are core and which are accessory. The traditional approach for generating a pangenome representation is to go through each individual genome and predict genes using our prior knowledge of what a gene ’looks’ like, before doing gene comparisons.
This prior knowledge might be biased in some way, meaning that the same gene shared between individuals might be predicted differently between genomes, even if we expect them to be the same. This inconsistency can hamper our efforts to analyse a pangenome – the same gene may not be identified in two different genomes, making a core gene look like an accessory gene.
Our work – ggCaller
We need a way of ensuring the same genes are predicted in the same way between genomes. To do this, we developed graph-gene-caller, also known as ggCaller. Rather than predicting genes in genomes one at a time, ggCaller predicts genes collectively within a network-like structure called a ‘graph’, which merges similar sequences between different genomes together. By scanning through the graph, ggCaller can identify genes shared between genomes, ensuring that similar genes are predicted in the same way.
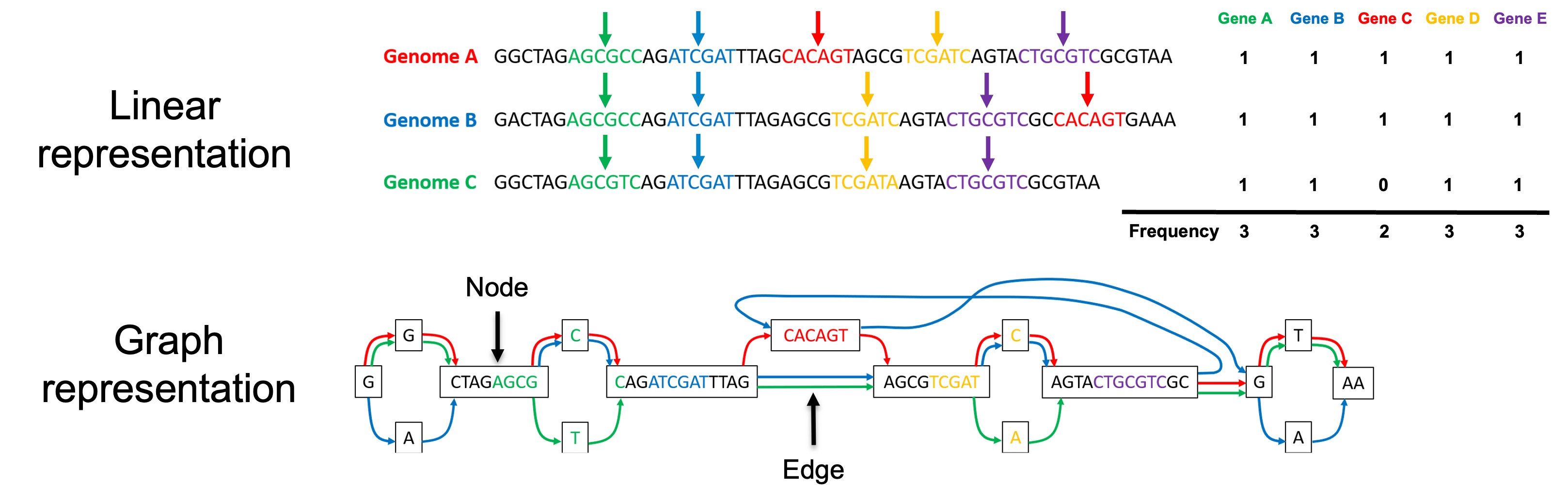
Comparison between predicting and comparing genes in linear genomes and a graph
ggCaller also annotates genes with functional labels. This is useful when you want to see not just how similar or different two members of the same species are based on their genes, but also what biological characteristics they share or differ by. By doing this collectively, we can share labels between the same genes between individuals, avoiding the need to re-label the same gene multiple times, saving time in the process.
How does ggCaller compare to existing tools?
We tested ggCaller on both simulated and real bacterial datasets to see how predicting genes collectively within a graph compares to predicting them in individual genomes. We found in many cases, ggCaller outperformed the traditional methods, with improved consistency in gene prediction across genomes. This increased consistency led to better estimates of the number of core and accessory genes present in a population.
We also tested ggCaller’s application in pangenome-wide association studies (PGWASs). These analyses look for statistical relationships between variants within a collection of genomes and a given biological characteristic. The problem with the existing methods, however, is that when you find a variant of interest, it is difficult to map it back to the original genomes, and therefore get a functional label for the variant. Using ggCaller, we show that using consistent gene prediction and annotation makes functional labelling, and therefore interpreting the biological effects of variants, much easier.

Neighbour-joining tree for PGWAS of resistance to tetracycline (A) and erythromycin (B) antibiotics in pneumococcus
ggCaller is also fast – we show that it can run on several thousand genomes and is still faster than existing workflows. Another benefit is that you get a pangenome from a single command, going directly from your genome sequences to functionally labelled genes, clusters, phylogenetic trees and more! This means you no longer need a pipeline of two or more tools to do pangenome analyses.

Fold-speed up of ggCaller over linear-genome based pipeline on three bacterial datasets
Check out the code and the paper
With the improvements in speed and accuracy for generating pangenome representations, as well as simplifying the process of predicting and comparing genes into a single tool, we believe ggCaller will be of great interest to researchers studying bacterial epidemiology and evolution.
ggCaller is available under the MIT-license on Github. For an in-depth guide and tutorial, see our readthedocs page. To read more about ggCaller, check out our publication in Genome Research.