Publications
A list of some of the papers directly from our group, with some short explanations.
2024
CELEBRIMBOR: Pangenomes from metagenomes
Joel Hellewell, Samuel T Horsfield, Johanna von Wachsmann, Tatiana Gurbich, Robert D Finn, Zamin Iqbal, Leah W Roberts, John A Lees (2024). CELEBRIMBOR: Pangenomes from metagenomes bioRxiv
https://www.biorxiv.org/content/10.1101/2024.04.05.588231.abstract
CELEBRIMBOR makes pangenome (specifically frequency estimates) from metagenome data.
MAGs (metagenome assembled genomes) have missing fragments, which can be quantified using single copy marker genes e.g. checkM. This systematically underestimates gene frequency. Previous papers had suggested using a lower core threshold to fix this.
Here, we instead made a probabilistic model to use these to readjust gene frequencies to make them more accurate, allowing parameterised choices of how to do so, in the package core gene threshold. We then wrapped it all up in a nice snakemake pipeline CELEBRIMBOR (Core ELEment Bias Removal In Metagenome Binned ORthologs) which lets you go from MAGs to pangenome.
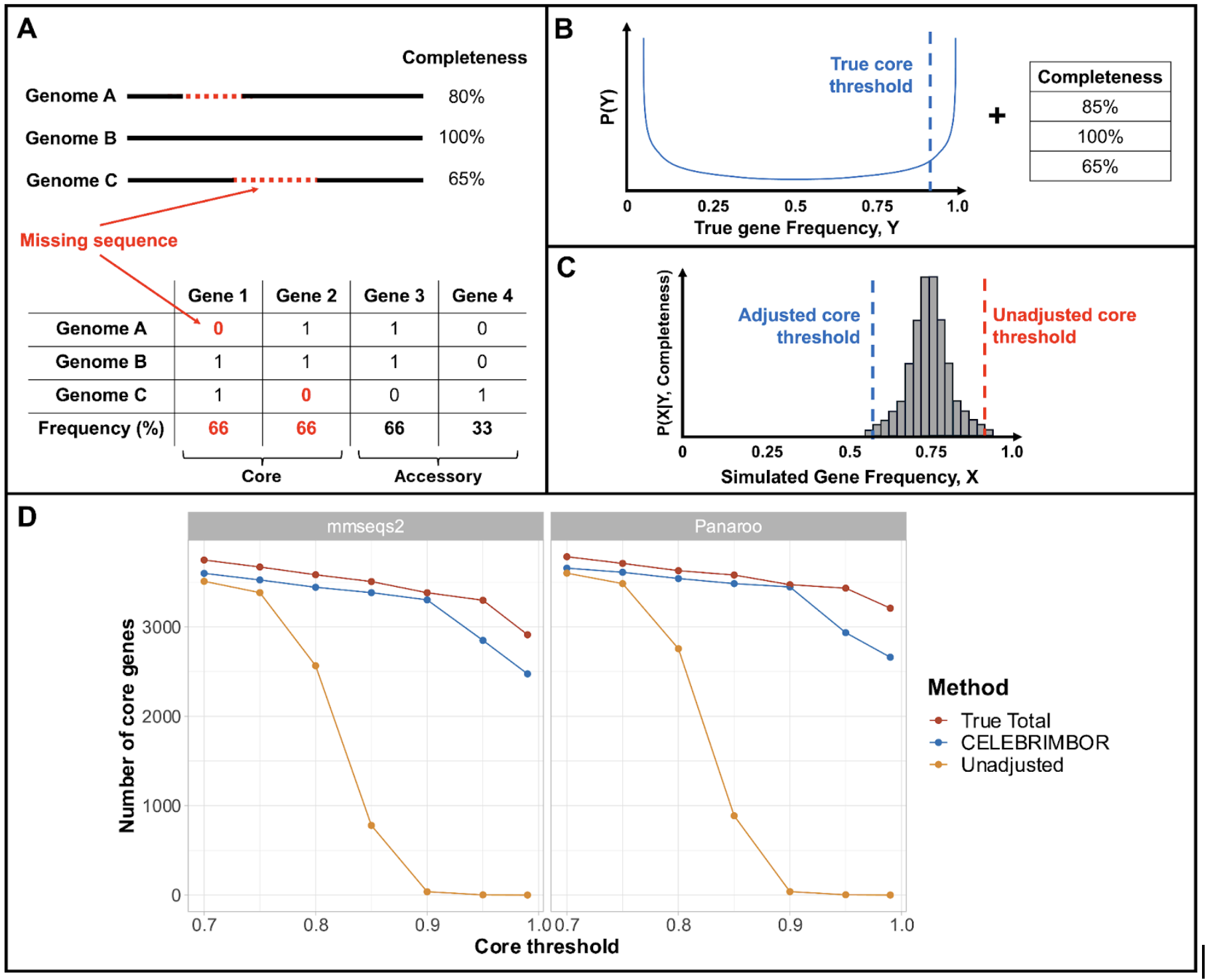
Seamless, rapid and accurate analyses of outbreak genomic data using Split K-mer Analysis (SKA)
Romain Derelle, Johanna von Wachsmann, Tommi Mäklin, Joel Hellewell, Timothy Russell, Ajit Lalvani, Leonid Chindelevitch, Nicholas J. Croucher, Simon R. Harris, John A. Lees (2024). Seamless, rapid and accurate analyses of outbreak genomic data using Split K-mer Analysis (SKA) bioRxiv
https://www.biorxiv.org/content/10.1101/2024.03.25.586631.abstract
This is the successor to SKA1, which uses split k-mers XXXX[-]XXXX to rapidly variant call microbial genomes, either reference-free or reference-based. SKA2 can be downloaded here.
We think that SKA is:
- The fastest, most accurate variant caller for microbial genomes.
- Has new ideas for nucleotide analysis in pangenomes, while avoiding reference bias.
- Accompanied by high quality software, will be useful for researchers from many different backgrounds.
If you’ve already been using it have a look at the figures to see benchmarking for when it is useful compared to other tools – very useful in outbreaks, still useful within strains (poppunk clusters, STs, CCs etc), drops off at the whole species level for most things. The code’s been available for a year already which has been a good opportunity for us to fix user reported bugs and add missing features!
Graph-based Nanopore Adaptive Sampling with GNASTy enables sensitive pneumococcal serotyping in complex samples
Samuel T. Horsfield, Basil Fok, Yuhan Fu, Paul Turner, John A. Lees, Nicholas J. Croucher (2024). Graph-based Nanopore Adaptive Sampling with GNASTy enables sensitive pneumococcal serotyping in complex samples bioRxiv
https://www.biorxiv.org/content/10.1101/2024.02.11.579857v1.abstract
Nanopore adaptive sampling can reject DNA during sequencing by reversing the voltage on sequencing pores. By making this decision dynamic and dependent on what has been sequenced (in the current pore and the sample as a whole) unwanted sequences can be depleted.
We tested and optimised an application to serotyping S. pneumoniae. This is based on a fairly long and structurally complex part of the chromosome, so PCR doesn’t work, but whole genome sequencing is usually overkill (or doesn’t pick up minority variants).
We showed that this technique 1) works, 2) has various improvements over alternatives such as microarray and physical methods (i.e. quellung or latex agglutination), 3) can be further improved by using graph-based references of the capsule operon. We called our technique GNASTy (Graph-based Nanopore Adaptive Sampling Typing, pronounced ’nasty’).
We also showed that adaptive sampling is much less effective at enriching for closely related samples, which has implications for its rollout in other applications of metagenomic sequencing.
Optimising machine learning prediction of minimum inhibitory concentrations in Klebsiella pneumoniae
Gherard Batisti Biffignandi, Leonid Chindelevitch, Marta Corbella, Edward J. Feil, Davide Sassera and John A. Lees (2023). Optimising machine learning prediction of minimum inhibitory concentrations in Klebsiella pneumoniae Microbial Genomics 10(3): 001222
https://www.microbiologyresearch.org/content/journal/mgen/10.1099/mgen.0.001222
Minimum inhibitory concentrations (MICs) are measured when collecting data on resistance to antibiotics. These are a semi-quantitative measurement (ordinal) giving the nearest doubling in drug concentration at which resistance appears.
In typical uses, clinical breakpoint guidelines are used to interpret these values into a dichotomised resistant or sensitive classificaition. However, these breakpoints can change over time and in some cases are propreitary. Furthermore, this classification loses information.
In this work, we looked at the performance of three typical methods used in genotype-to-phenotype prediction and inference in bacterial genomes: linear mixed models, elastic net regression and random forests. We analysed their performance with different binning resolutions and censoring of MIC data.
Using simulations and a real dataset of Klebsiella pneumoniae we are able to provide guidance for fitting these models, and collecting and reporting MIC data.
2023
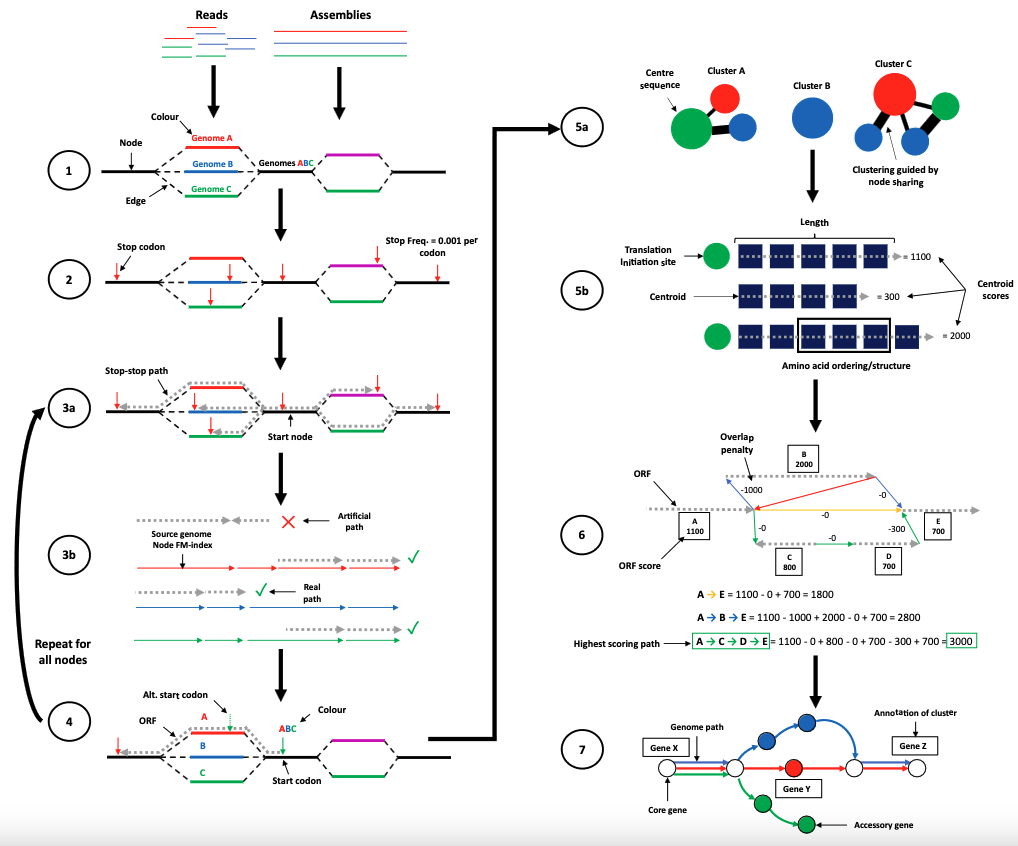
Accurate and fast graph-based pangenome annotation and clustering with ggCaller
Samuel T. Horsfield, Nicholas J. Croucher and John A. Lees (2023). Accurate and fast graph-based pangenome annotation and clustering with ggCaller Genome Research 33: 1622-1637
https://genome.cshlp.org/content/33/9/1622
Annotation of bacterial pangenomes is typically done one at a time. Each input assembly is annotated by calling and scoring open-reading frames (ORFs), and aligning all of these against databases to annotate function by homology. However, this is highly redundant. Many of these sequences will be shared and therefore the same computational operations used repeatedly. Furthermore, if genomes were annotated together population information could be used to guide this process, with higher quality sequences guiding annotation in lower quality samples, and overall more consistent gene calls.
ggCaller (graph-gene-caller) is a new approach which annotates ORFs within a population de Bruijn graph. Building on key tools bifrost and panaroo, ggCaller combines the steps of annotation and pangenome clustering, going from a set of input assemblies to annotated GFFs and a pangenome matrix. We show this saves time (up to 50x faster), and can give more accurate gene and cluster of orthologous genes (COG) calls, particularly in ‘difficult’ regions. We also show that this is a nice addition to the application and interpretation of bacterial GWAS. Good results can be obtained for whole genomes, but also smaller complex regions such as the capsule operon.
ggCaller packages a lot of the steps for analysis of bacterial genomes into one place:
- Annotation of fasta files to GFFs.
- Pangenome clustering and correction, typical plots (rarefaction, SFS).
- Core and accessory genome alignment.
- SNP calling in core and accessory genes.
- Structural variant calling.
- Sequence search.
- Basic phylogenetics and visualisation.
See the code on the software page, and also Sam’s blog post.
2022
Mandrake: visualizing microbial population structure by embedding millions of genomes into a low-dimensional representation
John A Lees, Gerry Tonkin-Hill, Zhirong Yang and Jukka Corander (2022). Mandrake: visualizing microbial population structure by embedding millions of genomes into a low-dimensional representation. Philosophical Transactions of the Royal Society B 377:20210237
https://doi.org/10.1098/rstb.2021.0237
Dimension reduction methods are a popular way to work with large amounts of genetic data: PCA, t-SNE and UMAP have all been used to analyse and visualise lots of samples in two-dimensions. These techniques are often used to cluster data, but have not been explicitly designed to do so. Some of these methods also stuggle to work with the millions of genomes now available.
Here we re-implement and extend the stochastic cluster embedding (SCE) method to work with genetic data, which is optimised to find visually identifiable clusters. Stochastic gradient descent is used to optimise, which we port to GPUs, and can run through datasets with millions of samples in a couple of hours.
We show good clustering of all bacterial data in the ENA as of 2018 (661k samples) and SARS-CoV-2 as of Nov 2021 (about 1M samples). We also make some fun videos and audio of the optimisation process.
See the code on the software page too.
Pneumococcal genetic variability in age-dependent bacterial carriage
Philip HC Kremer, Bart Ferwerda, Hester J Bootsma, Nienke Y Rots, Alienke J Wijmenga-Monsuur, Elisabeth AM Sanders, Krzysztof Trzciński, Anne L Wyllie, Paul Turner, Arie van der Ende, Matthijs C Brouwer, Stephen D Bentley, Diederik van de Beek, John A Lees (2022). Pneumococcal genetic variability in age-dependent bacterial carriage. eLife 11:e69244
https://doi.org/10.7554/eLife.69244
Previous studies have suggested that piliated S. pneumoniae strains are more commonly carried by infants, and there is a lot of evidence showing that infant immune response is different from adult immune response to carriage. Vaccine modelling studies have also suggested that to more effectively immunise against pneumococcal disease, the presently circulating population and distribution in adults and children should be accounted for.
We use genome-wide association studies (GWAS) to search for genetic factors associated with S. pneumoniae carriage in infants versus adults, in a meta-analysis over two populations. Overall we find wide between-cohort differences in strain composition, and between ages, but no clear independent genetic signals associated with infants or adults. This supports proposals future vaccination strategies which are primarily targeted at dominant circulating serotypes in specific pathogen populations.
Here’s our tree and metadata (view on microreact.org):