Visualising microbial population structure with mandrake
By John Lees
Paper: https://doi.org/10.1098/rstb.2021.0237
(Joint work with Gerry Tonkin-Hill)
Dimensional reduction and embeddings
Dimension reduction methods are a popular way to understand large amounts of genetic data: PCA, t-SNE and UMAP have all been used to analyse and visualise large numbers of samples in two-dimensions (with the latter being particularly popular with single cell techniques).
PCA is a linear transform into a coordinate system where the axes maximise the variance explained. t-SNE and UMAP are non-linear transforms which can better represent more complex underlying structure in the dataset, and are typically better at preserving local stucture (and generally do a better job of visualising everything in two dimensions, whereas in PCA higher axes may be needed). There’s a nice comparison of these methods by Huang et al, and a great interactive guide on the pitfalls on t-SNE by Wattenberg et al.
These techniques are probably best used in comparison with a labelling scheme (cell-type, for example), but can be used in an unsupervised context to cluster data. However, this isn’t what these methods are optimised for. On a more operational level, some of these methods also stuggle to work with the millions of samples now common in many datasets.
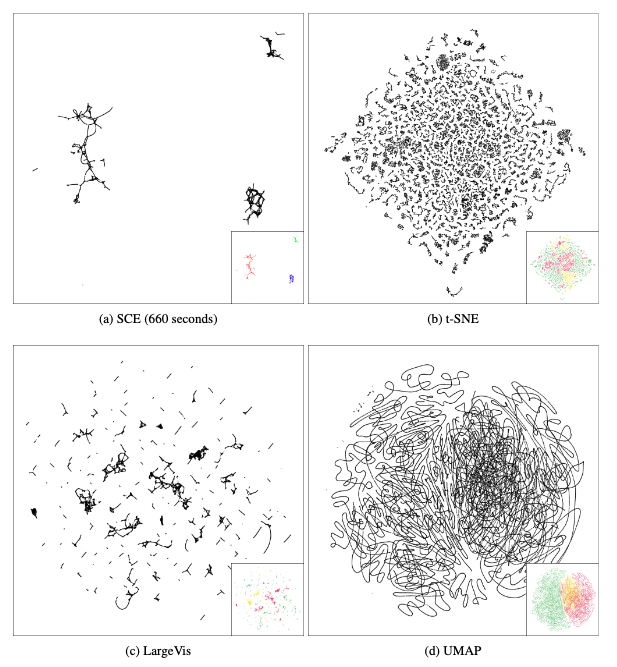
Visualizations of the TOMORADAR data set by different embedding methods
Our collaborators Zhirong Yang and Jukka Corander investigated this problem in a general context in their paper. They added a sliding parameter s which generalises the t-SNE approach.
Data can appear as more or less clustered, depending on the value of s. By asking a large number people whether data looked like it formed clusters, they were able to set a value that generally corresponds to human perception of clusters.
Additionally, they reformulated the problem as an objective function, which could then be solved by stochastic gradient descent, which is readily parallelisable, even running hundreds of thousands of threads on a GPU.
Our work – mandrake
In this paper, we further extended the method so we could apply it to genome datasets, calling the resulting tool mandrake:
- We linked with fast genetic distance esitmation from genes or SNPs with pairsnp, or from assemblies or reads using sketchlib.
- We automated and parallelised the entropy pre-processing step, needed with distance data.
- Automatically cluster the output using HDBSCAN.
- Added more plots of the output, including interactive versions.
- Re-implemented and optimised the CPU and particularly GPU (CUDA) code.
- Added a python API, so it is usable in other software.
- Created a WebAssembly version which you can run directly in the browser, without needing to upload data or install anything.
It’s all available as documented software available via conda so you can visualise and cluster your own genome assemblies, alignments or gene calls.
Running on genome datasets –- clusters at multiple resolutions
We demonstrated the method on four datasets. On simulated data from a coalescent model with recombination, run in a few different scenarios, mandrake was the only method consistently able to represent the underlying population structure.
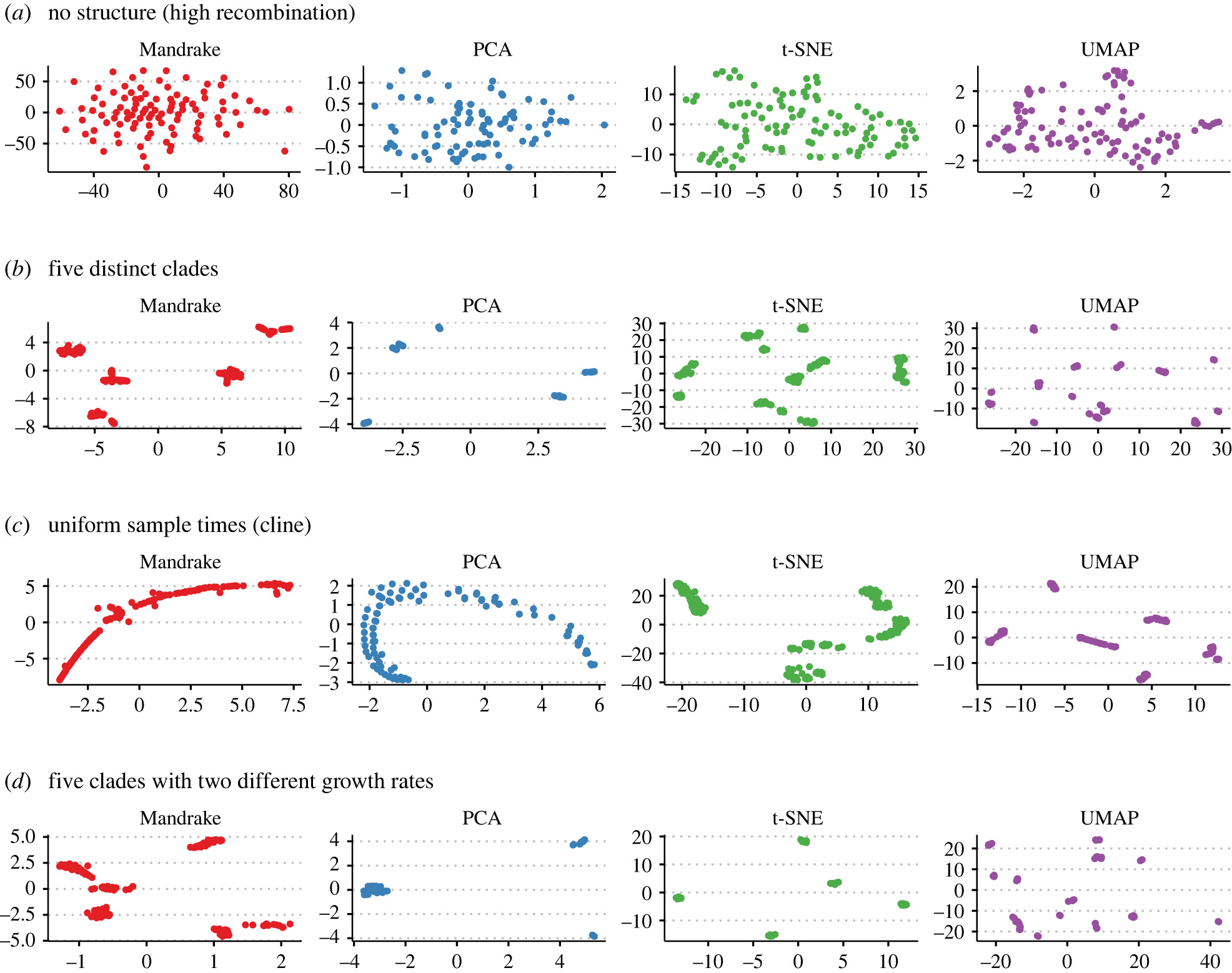
Embedding methods run on simualted genome data
The method scaled well, with datasets of 10k samples running in seconds. On the largest dataset tested aroof und one million SARS-CoV-2 genomes the embedding can be produced in about two hours (and finds the VOCs, without using these labels as input).
I think the most interesting example our run on 661k publicly available assemblies
from Blackwell et al.
There’s a video of this embedding being generated below (note you can generate this on your
own data by adding the --animate flag):
This is unlabelled data, and we just use nearest neighbour distances. Most species end up in their own cluster, which is nice to see. However we also found that some large collections like Salmonella form clusters of strains consistent with previous curated schemes. At the same time, due to its non-linearity, the same embedding is able to show separation of different lineages of Mycobacterium tuberculosis, despite their distances being a few orders of magnitude below species separation. We go into more detail of these results in the paper and the supplementary figures.
Give it a go!
We hope you like mandrake as a fast visualisation and clustering tool for a range of genomic data. I think it’s fun to see an explore all the output files, plots and animations on your own data, and would encourage you to try it out. This is even easier with the web app!
The API should also make it easy to put into python or C++ code, and we’ve already done this in PopPUNK v2.5.0.
Links
See the code on the software page too.
And make sure you check out the supplement to see all the videos!
You can find the paper here: John A Lees, Gerry Tonkin-Hill, Zhirong Yang and Jukka Corander (2022). Mandrake: visualizing microbial population structure by embedding millions of genomes into a low-dimensional representation. Philosophical Transactions of the Royal Society B 377:20210237. https://doi.org/10.1098/rstb.2021.0237
Code is here: https://github.com/bacpop/mandrake
Documentation is here: https://mandrake.readthedocs.io/en/latest/
A video showing some of these results:
What’s the bacronym?
A recursive acronym:
Mandrake Analyses Nucleotides, Dimension Reduces, Animates K-neighbour Embeddings