Peer Review of the pre-print 'Endonuclease fingerprint indicates a synthetic origin of SARS-CoV-2'
By Joel Hellewell
This is a peer review of the pre-print “Endonuclease fingerprint indicates a synthetic origin of SARS-CoV2”, it is highly recommended that you go and read the pre-print in order to understand this review.
Introduction
The broad thread of the argument in the pre-print is that a synthetically engineered COVID-19 virus would be created using a process where ‘restriction’ enzymes cut the vaccine genome into roughly equal fragments so that they can be cloned in a bacterial system before being reassembled. Restriction enzymes cut the genome at very select sites where the DNA matches certain short sequences. These target sequences, called restriction sites, may appear in the genome by chance as natural nucleotide mutations occur. However, for a given genome the restriction sites will not usually be in the equally spaced locations needed for cutting the genome into equal chunks using restriction enzymes. To get around this, scientists create mutations in the viral genome to create new, more equally spaced restriction sites and remove old restriction sites which are too close together. Observing equally spaced restriction sites might be a marker of a synthetically engineered virus, since a wild-type viral genome has a small probability of having restriction sites so equally spaced throughout the genome.
The authors construct a null distribution of the size of the largest DNA fragments that you would expect to see as a result of cutting genomes at restriction sites. They compare the largest fragment resulting from cutting a SARS-CoV2 genome with a certain pair of restriction enzymes to the null distribution and find that the largest fragment produced is unusually small. Under the null distribution there is under 1% probability of observing a largest fragment length that is equal to, or smaller than, the largest fragment length observed for SARS-CoV2. The authors therefore conclude that the “BsaI/BsmBI map of SARS-CoV-2 is anomalous for a wild coronavirus and more likely to have originated from an infectious clone designed as an efficient reverse genetics system”.
We argue that the issue with the argument in the pre-print is that it confuses the null hypothesis that it is actually rejecting, a claim that is strictly about the distribution of restriction sites across the genome, with a claim about the origin of the distribution of the restriction sites in the genome. The argument presented here is that the null distribution constructed in the pre-print gives the distribution of largest fragment sizes that we would expect to observe if restriction sites were uniformly distributed across the genome. That is, writing restriction site location in the genome as then so for all and elsewhere. Therefore, an observation having a low probability of belonging to this null distribution is evidence that restriction sites do not occur uniformly across the genome.
The rejection of this null hypothesis is not evidence that this lack of being uniformly distributed is due to a specific origin, namely scientists altering restriction sites. We argue that our understanding of how mutations occur naturally in a genome might lead to restriction sites locations that are not uniformly distributed. This is because locations in the genome where a restriction site can or is likely to emerge depends on the evolutionary history of the genome. Therefore, the viral genomes that we highlight here having a low probability of belonging to the null distribution constructed in the pre-print can be explained by a non-uniform distribution of restriction site locations occurring in genomes naturally.
Constructing the null distribution
In the pre-print the authors construct their null distribution by cutting up “a broad range of natural coronavirus genomes” using lots of different restriction enzymes with different restriction sites. This approximates the distribution of the length of the largest fragment sizes left after cutting genomes into a given number of fragments. There are 72 coronavirus genome samples which are each digested by 214 restriction enzymes, as well as a further 1000 randomly chosen pairs of the 214 restriction enzymes (out of 45,582 possible pairs). Since the 72 CoV samples are related, we expect the nucleotide sequences to be roughly similar, but we then cut up each sequence in 1214 different ways. This gives 87,408 samples in the null distribution, each one the result of digesting a given genome with a given enzyme or pair of enzymes.
We argue that constructing the null distribution in such a way gives an approximation of a general distribution for the distribution of largest fragment sizes occurring in genomes where restriction site locations are uniformly distributed across the genome. The null hypothesis in the pre-print is not specific for coronavirus genomes. Each time you cut the genomes with a new restriction enzyme you cut up the genome in a different way and measure the largest fragment. The more you do this, the more unique ways you cut up the genome and the more you approximate an equivalent distribution where you can simulate restriction site locations using the uniform distribution. After cutting any genome with enough different restriction enzymes that recognise enough distinct restriction sites you will produce approximately the same distribution (we try to express this visually in Figure 1). We will demonstrate this in two ways: i) showing that the null distribution in the pre-print is very similar to a distribution constructed explicitly using uniform distribution samples and ii) showing that the null distribution in the pre-print is also very similar to the distribution constructed by digesting the genome of a completely different organism using the same 1214 enzyme combinations.
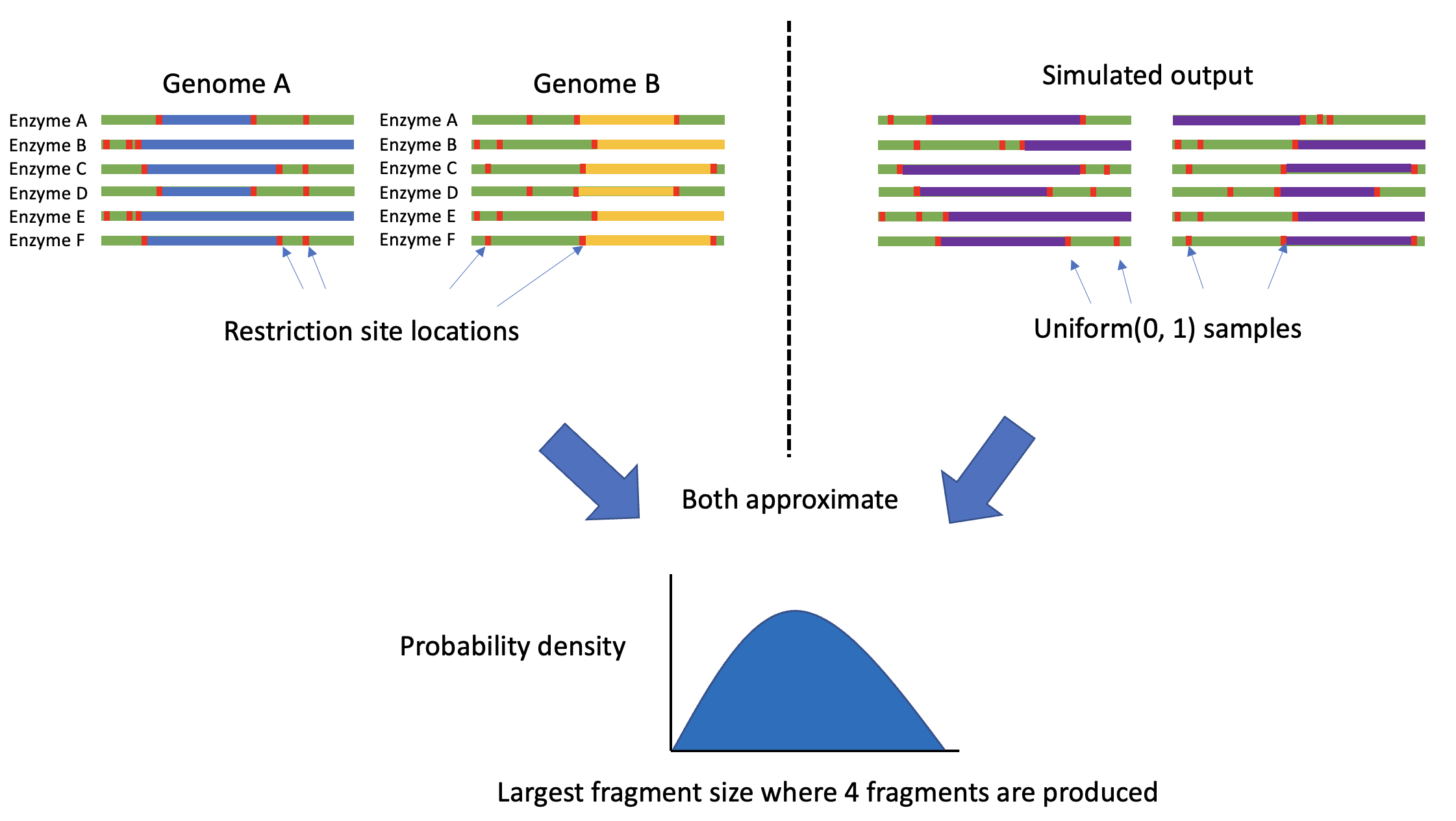
Figure 1: A schematic of how digesting multiple genomes with multiple restriction enzymes is an approximation of assuming that restriction sites are uniformly located within the genome.
Firstly, we will use samples from the uniform distribution to construct our own theoretical null hypothesis. If we wish to cut a genome into N fragments, we can take N draws from the uniform distribution between 0 and 1, order them, and then treat them as the location of restriction sites (right hand side of Figure 1). The biggest difference between our ordered samples is the largest fragment size as a proportion of the entire genome length. We can then look at the 95% quantiles across many simulations to give an approximation of the distribution of largest fragment length. We can plot this simulated distribution next to the null distribution from Figure 3C in the pre-print and see that they look very similar (Figure 2). Below is some R code that will quickly construct the desired distribution.
library(data.table)
# A vector for number of cuts into the genome, 10000 samples for each
# number of cuts
cuts <- rep(0:29, rep(10000, 30))
# We take n = cuts uniform samples, sort them, cap with 0 and 1
# so the vector is c(0, u1, u2, u3, ..., 1)
# then diff() gives a vector of fragment sizes
# and max() gives largest fragment size, which we return
longest_frag = vapply(cuts,
FUN = function(x){return(max(diff(c(0, sort(runif(x, 0, 1)), 1))))},
FUN.VALUE = c(1))
dt <- data.table(no_fragments = cuts + 1,
longest_frag)
# Calculates upper and lower 95% quantiles of max fragment size for
# each number of fragments
dt <- dt[, .(lq = quantile(longest_frag, 0.025),
uq = quantile(longest_frag, 0.975)), "no_fragments"]
To further prove that the null distribution constructed in the pre-print is not specific to coronavirus genomes, I took 10 randomly selected 30kb windows from the genome of the Eurasian red squirrel (Sciurus vulgaris) and digested them in-silico in the same manner as the pre-print. The resulting distribution for the red squirrel is very similar to both the distribution in the pre-print and the distribution explicitly utilising uniform location samples (Figure 2).
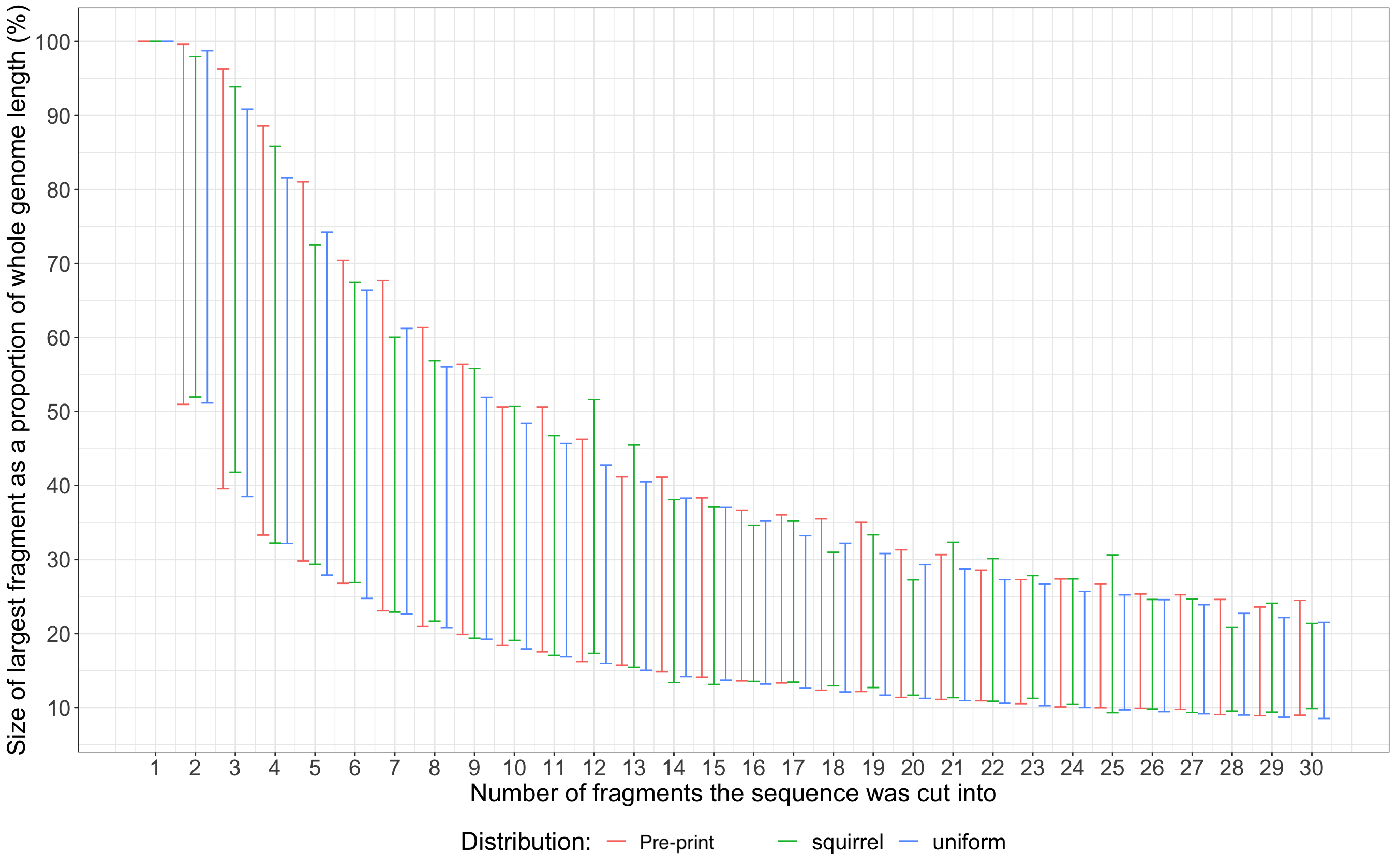
Figure 2: A comparison of three different largest fragment distributions. The null distribution from the pre-print (red), the distribution constructed in the same manner as the null hypothesis from a squirrel genome (green), and the distribution explicitly constructed using uniform distribution samples (blue).
The distributions will all be similar because they are all approximations of the distribution of largest fragments that result from the same data generating process - a process where the location of restriction sites is uniformly distributed across the genome. This is either because you explicitly use uniform samples or you approximate this process by digesting enough genomes with enough enzymes. Therefore, if an observation such as SARS-CoV2 has a low probability of belonging to this null distribution then the null hypothesis that you are rejecting is a hypothesis about the distribution of restriction site locations. This is specifically a claim about the distribution of restriction site locations and provides no evidence about the alternative distribution of restriction site locations or their origin, be it natural or synthetic, that led to your observation.
When constructing their null distribution the authors state they are calculating “which random distributions of restriction sites can be expected in non-modified viruses”, this is an unhelpful framing because it sets up opposition between the observation and the null distribution in terms of virus origin. But, as we have seen, observations differing from the null distribution is purely a matter of evidence for or against the distribution of restriction site locations across the genome. We argue that when looking at the distribution of fragment sizes for many restriction enzymes across many genomes, the result is approximately that restriction sites are uniformly located across the genome. However, for one restriction enzyme cutting one genome, the location in the genome where further restriction sites for this enzyme may emerge through mutation is a heavily structured process that depends on the current (and past) genome sequence and the possible mutations that can occur. This means that restriction site locations that emerge through a non-synthetic process of nucleotide evolution are not uniformly distributed across the genome and we would therefore expect many enzyme/genome pairs to produce largest fragment sizes with a low probability compared to the null hypothesis used in the pre-print.
Comparing observations to the null distributions
We now look at observed genomes with largest fragment sizes that have a low probability under the null distribution. As well as comparing data for SARS-CoV2 to the null distribution, we can also compare a magpie/robin CoV genome named HKU18 that is also included in the data for the pre-print. When cutting is performed using the enzyme “GC/TNAGC”, there are 6 fragments produced and the largest fragment is 87% of the length of the whole genome. Under the null distribution, the probability of seeing a largest fragment of this size or larger is ~0.03%. This sample is an outlier in the null distribution but in the opposite direction because the largest fragment is much larger than expected. Should the same inference about the origin of HKU18 be made as it has been for SARS-CoV2? Is the unusually big largest fragment a sign that restricted sites have been removed from HKU18 synthetically? In addition to HKU18, there is also a Erinaceus (hedgehog) CoV sample where the largest fragment produced using the enzyme “G/AATTC” is 24% of the size of the whole genome. This is even smaller than the largest fragment highlighted for SARS-CoV2 in the pre-print. Should the same conclusion about the origin of this sample be made as it has been for SARS-CoV2? Maybe scientists tried their synthetic coronavirus out on hedgehogs first?
These other samples pose a problem for the synthetic origin conclusion because they are allegedly wild-type viruses that also have a low probability under the null distribution. There are a few ways to try and overcome this problem. Firstly, you could argue that some of the samples are natural and some are synthetic. Perhaps the hedgehog and magpie CoV samples are examples of type 1 errors where you reject the null hypothesis even though it is true. But how do you work out which of these examples are type 1 errors or not without an a priori suspicion of the thing you are trying to prove, namely that SARS-CoV2 is synthetic. You could argue that your alleged fingerprint of synthetic origin is a necessary, but not a sufficient condition of being synthetic. However, this would make using the fingerprint as a marker of conclusive synthetic origin a case of affirming the consequent: If A, then B. B, therefore A. If a virus is synthetically engineered then it has unusual fragment sizes. SARS-CoV2 has unusual fragment sizes, therefore it is synthetically engineered. Another option available is to argue that all three examples are synthetic, but this seems far less plausible and would open you up to having to accept that any further discovered enzyme/genome combinations that have a low probability under your null hypothesis are also synthetic.
We argue that having a low probability of belonging to the null distribution is evidence for rejecting that the sample came from the data-generating process of the null distribution (i.e. that restriction site locations are uniformly distributed across the genome). A synthetic origin is one reason why a sample might not conform to this data-generating process, but it struggles to explain why CoV in hedgehogs also seem suspicious or why some samples might have unexpectedly large longest fragments. We would argue that an examination of the process of natural emergence of restriction site locations through random nucleotide mutations suggests that they are not uniformly distributed across the genome. This explanation can account for both unusually large and small largest fragments arising from restriction enzyme cutting.
How do new restriction site locations emerge naturally?
BsaI, one of the enzymes in question, cuts the genome at the location where it sees the specific sequence “GGTCTC”. Some regions of the SARS-CoV2 genome will almost match this nucleotide sequence already; it would take one mutation to make “GGTCTA” a restriction site and six mutations to make “TTGAGT” a restriction site. Making the simplifying assumption that nucleotide mutations happen at a constant rate across the genome, this already suggests a non-uniform structure to the emergence of new restriction sites in the genome. Restriction sites are more likely to emerge where the DNA sequence already closely resembles the sequence that BsaI requires. Below is a plot of how similar each 6-mer in a SARS-CoV2 genome provided in the pre-print is to a restriction site (or its reverse complement). There are very clear regions of the genome where more mutations would be required for a new restriction site to emerge.

Figure 3: The number of mutations required to create a restriction site location (or its reverse complement) in each 6-mer of the genome 'SARS2-WHu1' with accession number NC-045512. Numbers in red show the number of 6-mers in each group.
However, there is another layer of complexity in the locations where restriction sites may emerge. Viral genomes are composed of separate genes, many of which will be instructions for assembling amino acids into the complex proteins required for the various biological functions that the virus performs. Through the process of transcription and translation the DNA relates to the amino acids in the protein in blocks of three nucleotides called codons. Some codons refer to the same protein, so one or more of the three nucleotides in a codon can mutate and the same acid will be added to the protein. This is known as a synonymous or “silent” mutation. Synonymous mutations are usually considered to be evolutionarily neutral, since they produce no changes in the protein composition (although recently it seems like there may be some selection occurring because of the different efficiencies of tRNA molecules).
If the nucleotides in a codon mutate and a different amino acid is added to the protein as a result, this can change the function of the protein and alter the workings of the virus (sometimes even in a way which benefits the virus). When the amino acid changes this is known as a non-synonymous mutation. Some non-synonymous mutations can interrupt the function of a protein in a way that makes the virus non-viable, it will not reproduce and this line of evolution will not continue. There are usually conserved regions of the genome where we observe very few non-synonymous mutations between different viral samples because most changes here are fatal to the virus. On the other hand, changes to specific proteins might confer an advantage to the virus. A good example of this is mutations in SARS-CoV2 spike protein leading to a reduced immune response from the human host because their immune system has learned to recognise a very specific pattern of amino acids which have now changed. The emergence of restriction site location at a given location will by heavily dependent on a combination of i) locations where the genome already resembles the pattern recognised by BsaI and ii) places where the necessary mutations can occur, either as synonymous mutations or non-synonymous mutations that do not disrupt virus function terminally.
Nucleotide mutations will occur individually in a constrained and cumulative manner over time. There will be many unobserved stages of evolution between two genome samples that we observe (Figure 4). There will be many mutations that occur that render the virus unviable or may come with some fitness cost so that it is outcompeted by other strains, these stages will likely not be observed. At each stage a few random nucleotides will mutate, but we will only ever observe random mutations that don’t confer a disadvantage big enough for this virus to slip out of the evolutionary race. Restriction site emergence in a genome is an incidental phenomenon occurring as a result of viral evolution. Some areas of the viral genome already closely resemble the restriction site sequence, but where mutations can and will occur is due to the separate process of viral evolution. Sometimes the mutations that occur through evolution will introduce a new restriction site, but we wouldn’t expect the distribution of these restriction sites for a given enzyme in a given genome to be uniformly distributed across the genome.
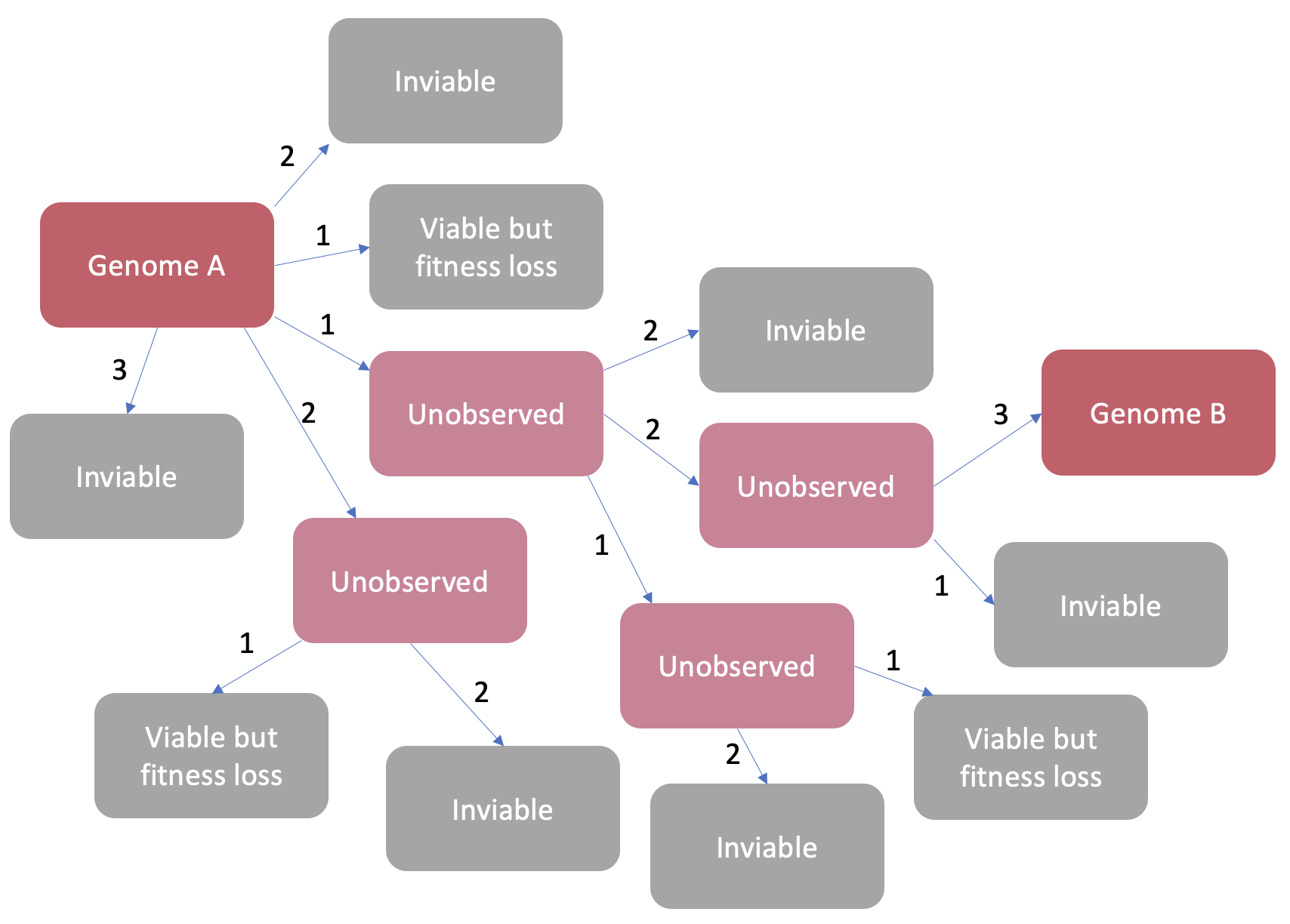
Figure 4: A schematic showing the process of accumulation nucleotide mutations between two observed genomes. There are many unobserved steps between each genome and many mutations caused terminal problems with virus function. The numbers on the arrows are the amount of nucleotide mutations between stages.
Mutation analysis
In the pre-print the authors simulate nucleotide mutations at random locations in viral genomes that are close evolutionary ancestors to SARS-CoV2, from this they conclude that the natural emergence of restriction sites in the locations where they are found in SARS-CoV2 is unlikely. The authors take the two most similar genomes to SARS-CoV2, add in 200-300 nucleotide mutations at random sites, and then digest them with a BsaI/BsmBI combination. They do this 100,000 times for both genomes and produce a distribution of maximum fragment size for a given number of fragments produced. Now, instead of digesting 72 different CoV genomes with 1214 enzymes they are digesting 200,000 very similar genomes with 1 enzyme.
The issue here is that the analysis ignores the constraints that evolutionary selection places upon mutations that might cause restriction site emergence. The mutation analysis is akin to saying that, in Figure 4 if we ignore viability, Genome B is unlikely to evolve from Genome A because there are so many other boxes that Genome A could end up as. How do we know that any of the mutant genomes generated by mutating 200-300 nucleotides in RaTG13 or BANAL-20-52 represent a viable virus? Our actual observations are heavily conditioned on natural selection washing away the many failed mutations and presenting us with only the ones that survive, no matter how contingent these particular mutations may seem in the huge space of possible nucleotide mutations that could have occurred. Saying that a SARS-CoV2 sample seems unusual compared to a batch of random silicon mutants that came into being outside the process of evolution and natural selection is not a convincing argument.
Conclusion
In conclusion, our main arguments against the analysis in pre-print are as follows:
- The location where restriction sites are located for a particular restriction site enzyme in a particular genome is not likely to be uniform. Where these sites will emerge is determined by an interplay of existing sequence similarity to the restriction enzyme target sequence and where such mutations are possible without negative consequences for the virus.
- The null distribution constructed in the pre-print, constructed by digesting many coronavirus genomes with many restriction enzymes, is the same as explicitly assuming that restriction sites locations are uniformly distributed throughout the genome.
- Therefore, samples having a low probability of belonging to the null distribution in the pre-print is evidence about the suitability of the uniform distribution assumption, not the origin of the distribution of restriction site locations.
- That restriction site locations emerge naturally, but not uniformly across the genome, is a better explanation for many other samples whose observation has a low probability under the null distribution. It can also explain why some observations have unusually large fragments left after cutting with restriction enzymes.
- The mutation analysis in the pre-print is not a good way of assessing the probability of restriction site locations emerging in an ancestor of SARS-CoV2 because it ignores that mutations may confer some selection advantage or disadvantage on the evolving virus.